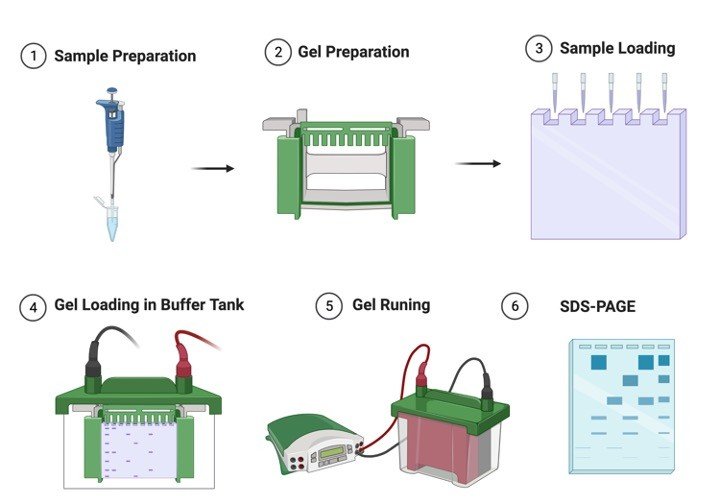
1. Acrylamide and N,N’ -methylene bisacrylamide: Prepare a stock solution containing 29% (w/v) acrylamide and 1% (w/v) bisacrylamide in deionized warm, water (to assist the dissolution of bisacrylamide).
Note: Acrylamide and bisacrylamide are slowly converted to acrylic and bisacrylic acid upon storage.
The reaction is catalyzed by light and alkali.
Hence always check that the pH of the solution is 7.0 or less, and store it in dark bottles at room temperature.
The fresh solutions should be prepared every few months.
Caution: Both are neurotoxins.
Polyacrylamide is considered to be non-toxic but care is to be taken while handling it as it might contain some amount of unpolymerized material.
2. Sodium dodecyl sulphate (SDS): Prepare a 10% (w/v) stock solution in Deionized water and store at room temperature.
3. Tris buffers for the preparation of resolving and stacking gels: Prepare 1.5 M Tris Cl. (pH 8.8) for resolving gel and 1.0 M Tris.Cl (pH 6.8) for stacking gel.
4. TEMED (N,N,N’N’-tetramethylenediamine): TEMED accelerates the polymerization of acrylamide and bisacrylamide by catalyzing the formation of free redicals from ammonium persulfate.
5. Ammonium persulfate (APS): It provides free radicals for polymerization of acrylamide and bisacrylamide.
Ammonium persulfate decomposes slowly and fresh solution is to be prepared weekly.
Prepare a 10% stock solution in Deionized water and store at 4C.
6. Tris-glycine electrophoresis buffer: 25 mM Tris base, 250 mM glycine and 0.1% SDS.
Adjust pH 8.3.
Prepare a 5X stock by dissolving 15.1 g of Tris base and 94 g of glycine in 900 ml water.
Adjust pH 8.3.
Add 50 ml of 10% (w/v) stock solution of SDS.
Make up the volume to 1000 ml with water.
7. 2X SDS gel-loading buffer: 100 mM Tris Cl (pH 6.8), 10% β-mercaptoethanol, 4% SDS, 0.2% bromophenol blue, 20% glycerol.