Phenylketonuria is an inborn error of phenylalanine metabolism which is due to the decreased metabolism of the amino acid.
If left untreated the conditions may lead to seizures, intellectual disability, mental problems and behavioral disorders.
It also leads to lighter and musty smell skin.
A baby which was born to a mother who is heterozygous carrier of this disease also gets affected with small head and low weight at birth.
Cause of Phenylketonuria
Phenylketonuria is an inherited genetic disorder due to a mutation in a PAH gene which causes low levels of the secretion of an enzyme phenylalanine hydrolase.
This is generally an autosomal recessive condition where both the copies of gene have to be defected to acquire this condition.
Types of Phenylketonuria
There are two types of phenylketonuria namely classic PKU and variant PKU depending upon the function of the enzyme.
However, the individual with only one copy of mutant gene do not have any symptoms. In many countries the screening test is done to the new born to protect them from severe symptoms.
It affects 1 in 10,000 people every year. However early screening can help to start the treatments quickly and helps in avoiding death of an individual in early ages.
Symptoms of Phenylketonuria
Phenylketonuria causes mild to severe ranges. The severe form of phenylalanine is known as classic variant.
If the baby is suffering from this classic variant it shows only mild symptoms and does show severe symptoms.
If it left untreated it leads to severe symptoms such as seizures, tumors, trembling and shaking of the limbs, stunted growth, hyperactivity, and also eczema in the skin and musty like odor in skin, urine and even in breath of an affected individual.
If Phenylketonuria is not diagnosed at an early stage it can also leads to irreversible brain damage and disabilities of intellectual abilities and also other behavioral problems in the adult children.
The other type of Phenylketonuria is also known as variant Phenylketonuria which does not cause any severe symptoms or Non Phenylketonuria hyperphenylalaninemia.
This occurs mostly in babies who has too much phenylalanine but they cause only mild symptoms in infants.
On following a proper diet from their Early stages Phenylketonuria can be prevented from becoming severe and also can be diminished.
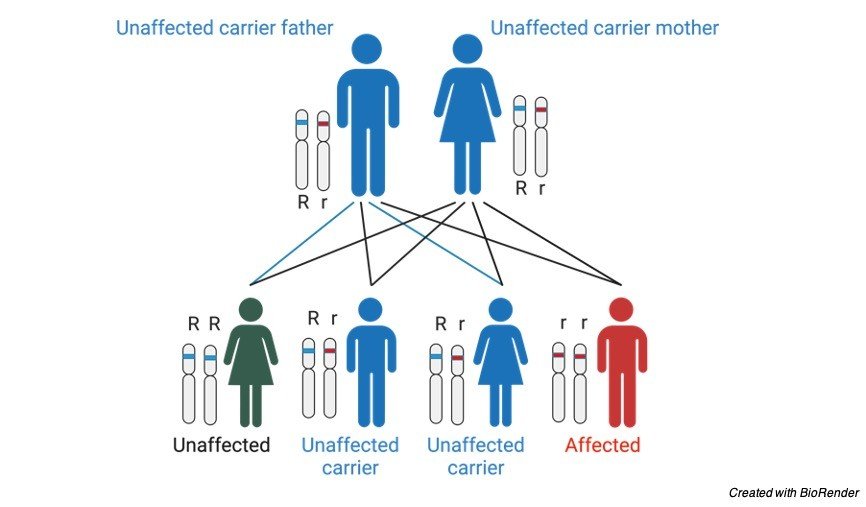
Pedigree Analysis of Phenylketonuria
As we know this condition is caused due to an autosomal recessive condition which means that where the parents are in a heterozygous condition of being a carrier, it affects the child.
The graph for this is listed below.
Frequency of Getting Phenylketonuria
The frequency of obtaining this disease ranges according to the geographical regions. Phenylketonuria occurs 1 in 10,000 to 15,000 babies; who are new born.
They are mostly detected at an early stage with the help of screening tests and treatments are started early, so the severity of this diseases can be prevented. So that the classic Phenylketonuria can be prevented from obtaining and variant form of this disease can be diminished.
Cause of Phenylketonuria
The genetic mutation that is happening in the PAH gene results in phenylketonuria. As PAH gene only instructs the body cells to synthesise an enzyme known as phenylketonuria; which helps in the conversion of amino acid phenylalanine to form other vital components that are needed for the body.
Due to this gene mutated condition, the activity of phenylalanine hydrolase is reduced which effects the activity of processing phenylalanine in diets.
This leads to a condition of building toxic levels in blood cells and other tissues of the body.
This condition occurs because the nerve cells in the brain are sensitive particularly to the phenylalanine levels where excessive production can cause damage to the brain cells.
Classic PKU happens when a phenylalanine activity is reduced to the extent levels or it may be absent at most severe cases.
People suffering from this condition if left untreated it causes severe brain damage and other serious health problems.
Mutation in this gene retains the activity of some enzymes and results in milder variant PKU.
Phenylketonuria Diagnosis
Screening test is one of the most popular test. It is performed in babies whose parents have this condition of inheritance.
Through this test it can be identified that whether the gene is inherited from parents to their children and if found in early stages it can be treated and cured before its s becoming severe.
These tests are done after 6 weeks of birth of a child.
Treatment of Phenylketonuria
The babies found out with this type of disorders can be provided or treated with proper nutritious diet which limits the intake of phenylalanine and they are led to breast fed to get the essential nutrients.
Physicians also provide these kinds of children a special formula of diet known as Lofenalac which is very high in protein.