Primary cultures denote those cultures which consist of cells before subculturing.
It represents the mixture of cell types present in a given tissue and hence they are closer to the tissue architecture in vivo.
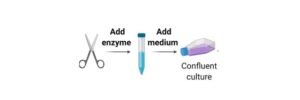
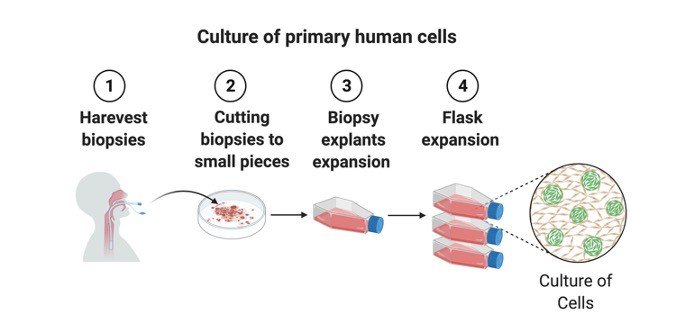
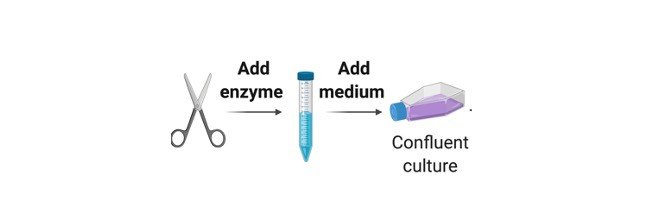
In primary cultures the tissue pieces can be grown as explants cultures or tissues can be subjected to enzyme treatment and dissociated cells are grown as monolayers.
Distinguishing Features of Organ Cell Culture
Architecture of tissues in vivo is retained.
Their functionality is also retained, since they maintain their differentiated character.
The tissue is cultured at the liquid-gas interface.
Grids, gels and sponge matrix which maintain 3D structure of organs are used as substrates.
Attempts are made to deliberately minimize proliferation and migration of cells.
Advantages of Organ Cell Culture
Organ cultures are used for studies involving cell interactions, functional properties of cells, effects of external agents or effects of products of one organ on another.
Characterization of these cultures can be easily carried out from histological studies.
Biochemical differentiation is also possible.
Disadvantages of Organ Cell Culture
1. Organ cultures cannot be propagated for long periods.
2. Each experiment requires a new culture.
3. Quantitation of results is very difficult.
4. High inter-sample variation is required for reproducibility and homogeneity of results.
Requirements of Organ Cell Culture
Unsterile-fertilized chick eggs incubated for 10 days.
Sterile-surgical instruments, 65 mm pertidishes, PBS-A with antibiotics, 35 mm petridishes, RPMI 1640 with 20% FCS, trypsin 50 ml conical flasks, magnetic bars.
Organ Cell Culture Procedure
1. Wipe precleaned eggs with savlon and iodine.
2. Hold the egg with broad side up and tap the shell gently with scalpel to crack it uniformly all around.
3. With a sterile forceps gently remove the shell pieces and displace the CAM to expose the embryo.
4. Lift up the embryo by inserting one arm of a blunt forceps below the neck of the embryo and transfer it to a petridish with PBS-A.
5. Wash the embryo twice with PBS-A, decapitate and wash again twice.
6. Dissect out all the visceral organs wash the remaining parts of the embryo with PBS-A, mince it with sharp scissors and transfer to a 50 ml conical flask.
7. Add 10-15 ml trypsin, put a magnetic bar in the flask and keep the flask on the magnetic stirrer with a gentle speed to avoid frothing for 30 mins.
8. Remove the flask from the stirrer, allow the pieces to settle, transfer the supernantant to a centrifuge tube and centrifuge for 10 mins at 1500 rpm.
9. If necessary, repeat the step of trypsinization once or twice more.
10. Pool all the cell suspensions, make a cell count and add about 1 X 10^5 cells/ml in 35 mm petri dishes and incubate in the CO2 incubator.
Observation: Within 24 hrs cells form monolayer and show semi confluency in 4-5 days.
Primary culture is the culture of cells directly obtained from animal tissues.
It has the following advantages:
More in vivo like characteristics
Cells can be obtained from rare or unusual species
Patient derived tissues can be utilized for direct experimentation
Cells that are not available as cell lines
About Rat Peritoneal Macrophages Cell Culture
The two most convenient sources of primary murine macrophages are the bone marrow and the peritoneal cavity.
Resident peritoneal macrophages can readily be harvested from mice and purified by adherence to tissue culture plastic.
The injection of thioglycollate broth into the peritoneal cavity produces an inflammatory response allowing the purification of large numbers of elicited macrophages.
The macrophages obtained by the following protocol are normally 90% pure and can be used further for other assays like drug uptake, phagocytosis etc.
Cell Culture Requirements
Sterile:
1. Phosphate Buffer Saline (PBS) (i.e. 137mM NaCl, 2.7mM KCl, 10mM Na2HPO4.2H2O and 2 mM KH2PO4, pH 7.4).
2. Geys solution: Stock A (0.65M NH4Cl, 0.024 M KCl, 10.56 mM Na2HPO4, 0.89 KH2PO4, 0.02 M Glucose, 0.141 mM Phenol Red); Stock B (0.207 mM MgCl2.6H2O, 0.057 mM MgSO4.7H2O, 0.306 mM CaCl2); Stock C (2.68 mM NaHCO3); Geys Solution (Make Fresh): 20 Parts of Stock A 5 Parts of Stock B 5 Parts of Stock C 70 Parts of autoclaved deionized water.
3. RPMI medium supplemented with 10% FBS and antibiotics
4. FBS
5. 23G and 18G needles
6. 10ml syringe
7. 3% w/v Brewer’s complete thioglycollate broth: 3g dissolved in water, Autoclave and store in dark and should be allowed to age for atleast 15 days before using for the experiment
8. Scissors and forceps
Non-sterile:
9. 70% v/v isopropyl alcohol or ethanol
10. Cotton
11. Ice
Cell Culture Procedure
1. Inject the rat with 5 ml aged thioglycolate medium in the peritoneal cavity using a syringe with 27 G needle.
2. After 72 hr and immediately before harvesting the macrophages, sacrifice the rat by CO2 asphyxiation.
3. Disinfect all the external areas of the rat with 70% IPA.
4. Make a small incision in the abdominal region and gently rip the skin downward to expose the intra-peritoneal cavity.
5. Inject 10 ml of sterile PBS into the intra-peritoneal cavity using a syringe with a 23 G needle and massage the abdomen gently for 2 min.
6. Retract the injected PBS using a syringe with 18 G needle. Care should be taken not to injure the internal organs during this process.
7. Centrifuge the Macrophage suspension at 1300 rpm, 7 min and 40C and observe the pellet.
Note: If RBCs are visualized, then add 1ml of complete Gey’s solution and incubate on ice for 2-5 minutes. Gently overlay 1ml of FBS to this suspension and spin at 300g for 10 minutes at 4oC. Wash once with PBS containing 5% FBS and then resuspend in 1 ml of RPMI supplemented with FCS and antibiotics.
8. Count the cells using hemocytometer
9. Plate the resident peritoneal cells RPMI culture medium at 3 x 10^5 cells per well in a 24-well tissue culture plate or 106 cells in 30mm sterile petridish and incubate for 60 mins at 37°C.
10. Remove the nonadherent cells by washing 3 times in 500ml warm PBS, using a gentle swirling action.
11. The adherent cells normally contain >90% macrophages
12. These macrophages will be used for drug uptake assay
In the development and maintenance of the nervous system there is a complex interdependency between neurons and glial cells.
This relationship is vital for their individual differentiation, development, and functionality but also seems to play an important role in progressive neurodegeneration and in the modulation of neurotoxic effects.
Co-cultures of different cells of the nervous system (i.e., neurons, astrocytes, and microglia) represent the easiest approach to:
1) study intercommunication between the different populations of the nervous system,
2) evaluate its relevance in several physiological responses and/or propagation of the damage,
3) study the molecular mechanisms involved. Other possible approaches are to use aggregate cultures of neural and glial cells and the more complex organotypic slices of hippocampus.
Created with BioRender
Requirements for Cortical Neuronal Cell Culture
Sterile:
1. DMEM-F12 medium
2. B-27 Supplement
3. L-Glutamine
4. HBSS
5. FBS
6. Horse Serum
7. Trypsin (0.1%w/v in sterile calcium free PBS)
8. Cytosine Arabinoside (1mM stock in sterile PBS)
9. Antibiotic mixture
10. Sterile 24 well plates
11. Dissection instruments like scissors, fine forceps, needle, blade
12. Cotton
13. 15ml tubes
14. 0.5ml microfuge tubes
15. Tryphan blue (0.5% w/v in sterile Calcium and magnesium free HBSS)
16. 1ml tips, 200μl tips
17.
Non sterile:
17. Time mated 18 day pregnant female Swiss albino mice
18. Diethyl Ether (for anaesthetizing the animal)
19. 70% alcohol
20. Bench top refrigerated centrifuge
21. Water bath at 37 C
Cortical Neuronal Cell Culture Procedure
1. Neuroglial cultures are obtained from 16 to 18 days old embryonic SA mouse.
2. Anaesthetize the pregnant mouse with ether and remove the fetuses from the uterine horns. Place them in a sterile 60-mm petri dish with ice-cold HBSS.
3. Remove the fetuses from the amniotic sac and remove the brain of the fetuses.
4. Remove the cerebral cortices from the brain and place in ice cold HBSS with Ca and Mg
5. Mince the Cerebral cortices and enzymatically dissociate with 0.5% trypsin in HBSS without Ca++ and Mg++ for 20 min. at 37 ̊C on water bath in 15 ml centrifuge tube.
6. After the enzymatic dissociation mechanically dissociate the cells by means of 1ml pipette. Resuspend the dissociated cells into 2ml of DMEM-F12 medium with 10% Horse serum and 10% FBS
7. Count the number of viable cells using hemocytometer and tryphan blue exclusion method. The number of viable cells should not be less than 80-90%.
8. Plate the cells on to the 6/24 well plate at a density 1×10 – 3×10 cells per well in a culture medium containing DMEM-F12 supplemented with 10% Horse serum
9. After 48 hrs of the plating add Ara C (Cytosine arabinocyde 10μM final concentration). Addition of Ara C will help neurons to out compete in growth from glial cells
10. 24 hrs later remove the existing medium and add fresh DMEM-F12 medium with 10% Horse Serum and then every 3-4 day.
11. All the experiments can be carried out on the confluent cells (After 7-10 days.)